




During the Myopain Seminars dry needling courses, our instructors may discuss the possible role of dry needling with individual patients diagnosed with Ehlers-Danlos Syndrome (EDS). However, frequently our students are not familiar with EDS and some of the common features and comorbidities of the syndrome [1], which we will briefly review in this blog. We will discuss the possible role of dry needling in a future blog.
The EDS syndromes are a heterogeneous group of heritable connective tissue disorders featuring joint hypermobility, skin hyperextensibility, and tissue fragility. The most common types of EDS are classic and hypermobile EDS, which are two of the thirteen defined phenotypes of EDS [2]. The hypermobile EDS (hEDS) type represents at least 80%–90% of all EDS cases [3] and most patients with hEDS have significant levels of disability, including physical and psychological disability factors [4]. The altered structure of the connective tissues is the main culprit leading to widespread joint pain [5-7], neuropathic pain [8], fatigue [7, 9], gastrointestinal dysfunction [10], and even less obvious symptoms, such as poor responses to topical applications or injections of analgesic medications [11]. Patients with EDS report difficulty getting numb during dental procedures or staying anesthetized during surgeries.
Hypermobile EDS (hEDS) is the only type without a known genetic basis. Most EDS patients have heterozygous mutations in one of the genes encoding type V collagen (COL5A1 and COL5A2) [12]. The other types of EDS include vascular EDS, cardiac-valvular EDS, arthrochalasia EDS, dermatosparaxis EDS, kyphoscoliotic EDS, Brittle Cornea syndrome, spondylodysplastic EDS, musculo-contractural EDS, myopathic EDS, and periodontal EDS. Some types of EDS overlap with other clinical conditions, such as Brittle Cornea syndrome and myopathic EDS. Within the scope of physical therapy practice, it is very rare to see patients with these rare forms of EDS.
Clinicians working with patients diagnosed with EDS may perform the Beighton tests, which are reliable but have inconclusive validity [13]. It also does not capture everyone with EDS, partially because the hypermobility can be local, effecting only a few joints, or more widespread.
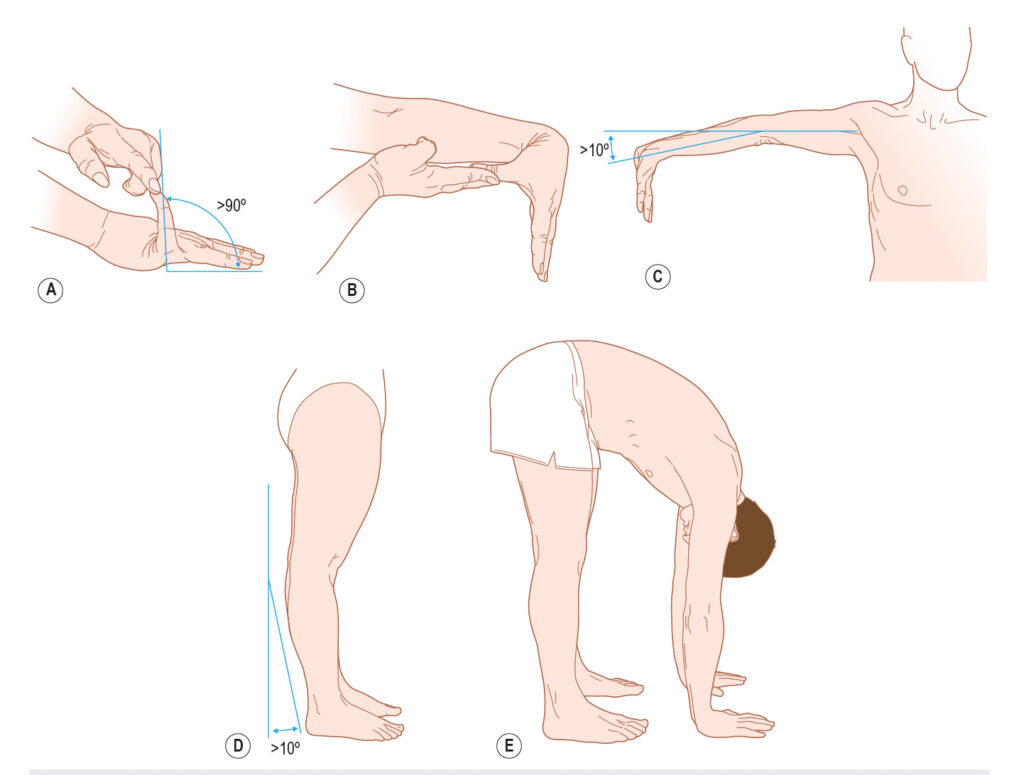
We also use the Five-Part Questionnaire, which is the most used questionnaire for adults with conflicting evidence of reliability and validity [13, 14]:
- Can you now (or could you ever) place your hands flat on the floor without bending your knees?
- Can you now (or could you ever) bend your thumb to touch your forearm?
- As a child, did you amuse your friends by contorting your body into strange shapes or could you do the splits?
- As a child or teenager, did your shoulder or kneecap dislocate on more than one occasion?
- Do you consider yourself “double-jointed”?
A “yes” answer to two or more questions suggests joint hypermobility with 80–85% sensitivity and 80–90% specificity.
As a multisystem disorder, EDS frequently coexists with a range of comorbidities that can significantly impair the quality of life.
 During the initial interview with patients suspected of having EDS, clinicians must explore which other problems may co-exist with the syndrome, including cervical instability, Chiari malformation with or without tethered cord syndrome [15], anterior brainstem compression [16], gastrointestinal disorders [17], dysautonomia [18], and mast cell disorders [19]. Another way to look at this is to rule out possible neurological complications resulting from tissue weakness, biophysical deformative stresses, entrapments, and tissue deformations [20].
During the initial interview with patients suspected of having EDS, clinicians must explore which other problems may co-exist with the syndrome, including cervical instability, Chiari malformation with or without tethered cord syndrome [15], anterior brainstem compression [16], gastrointestinal disorders [17], dysautonomia [18], and mast cell disorders [19]. Another way to look at this is to rule out possible neurological complications resulting from tissue weakness, biophysical deformative stresses, entrapments, and tissue deformations [20].
Cervical instability
Cervical hypermobility is a frequent manifestation of EDS. This condition leads to instability in the cervical spine because of the incompetence of the alar ligament, including atlantoaxial instability and rotary subluxations [21], anterior brainstem compression, and Chiari malformation [22]. Symptoms may include chronic neck pain, cervicogenic headaches, pre-syncope, syncope, lightheadedness, nausea, joint pain, numbness and weakness in the upper and lower extremities, and exercise tolerance [21]. Advanced imaging is required to diagnose cervical instability, including an upright MRI and rotational CT scan of C1-2 to show pathological angular displacement or lateral neck tilting to demonstrate lateral displacement [21, 23].
Chiari malformation
Chiari malformation involves the downward displacement of the cerebellar tonsils into the foramen magnum. This condition is particularly common in EDS patients due to the instability of the craniocervical junction and the inherent laxity of supportive tissues.
Symptoms of Chiari malformation often overlap with other EDS-related conditions. They may include severe occipital headaches, especially when coughing or straining, balance and coordination difficulties, nausea, dizziness, vertigo, Tarlov cyst syndrome, tethered cord, and impaired cerebrospinal fluid (CSF) flow, which may lead to syringomyelia [23-25]. The convergence of connective tissue disorders and complicated forms of Chiari are well recognized and characterized by basilar invagination, a kyphotic clivo-axial angle [25], and craniocervical instability [22].


Management of Chiari malformation typically involves neurosurgical decompression and correction of the clivo-axial angle to alleviate symptoms and prevent further neurological damage [22].
Anterior brainstem compression
Anterior brainstem compression occurs when structural instability in the craniocervical junction results in pressure on the brainstem. In EDS, ligamentous laxity and joint hypermobility can lead to the forward displacement of the odontoid process, compromising the brainstem’s structural integrity. The diagnosis is made by advanced imaging, especially an upright cervical MRI with determination of the Grabb-Oakes measurement and clivo-axial angle [23]. To determine the Grabb-Oakes measurement, draw a line from the basion to the posterior-inferior body of C-2. Measure the perpendicular distance (*) from that line to the dura.

A Grabb-Oakes measurement of 9 mm or more is indicative of anterior brainstem compression, which can lead to a wide range of symptoms, including, but not limited to occipital headaches, dizziness and vertigo, dysphagia, dysautonomia, incontinence, and lack of motor control, especially in the pelvis and lower extremities [16, 21-26].
Urinary incontinence
In a small study, Carley and Schaffer (2000) found that urinary incontinence and pelvic organ prolapse were common among women with EDS, which supports that connective tissue disorders may be associated with EDS [27]. Patel and Khullar (2021) confirmed that many pelvic symptoms are linked to EDS, including vaginal prolapse, overactive bladder symptoms, voiding dysfunction, bladder pain syndrome, recurrent urinary tracts infections, stress urinary incontinence, recurring bladder diverticula, vesicoureteral reflux, pelvic floor pain or spasms, and complicated postnatal perineal wounds [28]. A 2024 literature review estimated the prevalence of urinary incontinence among EDS patients at 50%-60% and 29%-75% for pelvic organ prolapse [29]. In the clinic, we have seen patients with a sudden onset of urinary incontinence along with other symptoms of anterior brainstem compression.
Gastrointestinal disorders
Gastrointestinal (GI) symptoms are highly prevalent in EDS and often stem from the fragility of the GI tract’s connective tissues. Because the stomach and intestine continue to stretch when filled with food fecal matter, patients with EDS often present with gastroparesis and a dysfunctional defecation reflex. In the stomach, this leads to a delay in stomach emptying, which contributes to nausea, vomiting, bloating, and early satiety. In the larger intestines, this causes symptoms of irritable bowel syndrome with complaints of chronic abdominal pain, diarrhea, constipation, and decreased motility. Furthermore, patients with EDS are more prone to experience hernias and rectal prolapse, and esophageal dysmotility and reflux, whereby esophageal connective tissues can cause difficulty swallowing and gastroesophageal reflux disease (GERD).
Dysautonomia
Dysautonomia, or autonomic nervous system dysfunction, is a common comorbidity in EDS. One of the most frequently observed forms is postural orthostatic tachycardia syndrome (POTS), characterized by an exaggerated heart rate response upon standing. Common symptoms include lightheadedness and fainting, cardiac palpitations and tachycardia, fatigue, and exercise intolerance, sudomotor dysfunction and temperature regulation issues, and gastrointestinal symptoms like nausea and bloating [18, 30]. De Wandele and colleagues (2014) concluded that neuropathy, connective tissue laxity, and vasoactive medication likely play a role in the development of dysautonomia in hypermobile EDS.
Mast Cell Disorders
Mast cell activation syndrome (MCAS) is another comorbidity frequently associated with EDS [31]. It involves the overactivation of mast cells and the release of inflammatory mediators like histamine, prostaglandins, and cytokines [18, 19]. Symptoms of MCAS vary widely but commonly include flushing, itching, and hives, gastrointestinal distress, respiratory symptoms, including wheezing and throat tightness, and cardiovascular symptoms, such as low blood pressure or anaphylaxis-like episodes.
Summary
The comorbidities of EDS are often interconnected, creating a complex web of symptoms that exacerbate one another. For example, craniocervical instability can worsen dysautonomia by affecting brainstem function, while gastrointestinal dysmotility can amplify symptoms of dysautonomia and mast cell disorders. This interplay necessitates a multidisciplinary approach to care, with collaboration among specialists such as neurologists, gastroenterologists, geneticists, and immunologists. Unfortunately, for many patients coming to Bethesda Physiocare, our physical therapy clinic in Bethesda, MD, the physical therapist is often the first healthcare provider familiar with the many comorbidities and possible solutions. Bethesda Physiocare is one of only 22 centers worldwide recognized as a Center of Excellence by the international EDS Society.
A few select references
- Dommerholt, J. and N. Mayberry, Hypo- and Hypermobility, in Fascia in Sport and Movement, R. Schleip, J. Wilke, and A. Baker, Editors. 2021, Handspring Publishing: Pencaitland. p. 77-95.
- Malfait, F., et al., The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet, 2017. 175(1): p. 8-26.
- Tinkle, B., et al., Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am J Med Genet C Semin Med Genet, 2017. 175(1): p. 48-69.
- Scheper, M.C., et al., Disability in Adolescents and Adults Diagnosed With Hypermobility-Related Disorders: A Meta-Analysis. Arch Phys Med Rehabil, 2016. 97(12): p. 2174-2187.
- Voermans, N.C., et al., Pain in ehlers-danlos syndrome is common, severe, and associated with functional impairment. J Pain Symptom Manage, 2010. 40(3): p. 370-8.
- Voermans, N.C., et al., Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol, 2009. 65(6): p. 687-97.
- Murray, B., et al., Ehlers-Danlos syndrome, hypermobility type: A characterization of the patients’ lived experience. Am J Med Genet A, 2013. 161A(12): p. 2981-8.
- Voermans, N.C., H. Knoop, and B.G. van Engelen, High frequency of neuropathic pain in Ehlers-Danlos syndrome: an association with axonal polyneuropathy and compression neuropathy? J Pain Symptom Manage, 2011. 41(5): p. e4-6; author reply e6-7.
- Voermans, N.C. and H. Knoop, Both pain and fatigue are important possible determinants of disability in patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil, 2011. 33(8): p. 706-7.
- Beckers, A.B., et al., Gastrointestinal disorders in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: A review for the gastroenterologist. Neurogastroenterol Motil, 2017. 29(8).
- Arendt-Nielsen, L., et al., Insufficient effect of local analgesics in Ehlers Danlos type III patients (connective tissue disorder). Acta Anaesthesiol Scand, 1990. 34(5): p. 358-61.
- Bowen, J.M., et al., Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet, 2017. 175(1): p. 27-39.
- Juul-Kristensen, B., et al., Measurement properties of clinical assessment methods for classifying generalized joint hypermobility-A systematic review. Am J Med Genet C Semin Med Genet, 2017. 175(1): p. 116-147.
- Hakim, A.J. and R. Grahame, A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract, 2003. 57(3): p. 163-6.
- Milhorat, T.H., et al., Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine, 2007. 7(6): p. 601-9.
- Henderson, F.C., Sr., et al., Utility of the clivo-axial angle in assessing brainstem deformity: pilot study and literature review. Neurosurg Rev, 2018. 41(1): p. 149-163.
- Botrus, G., et al., Spectrum of Gastrointestinal Manifestations in Joint Hypermobility Syndromes. Am J Med Sci, 2018. 355(6): p. 573-580.
- Bonamichi-Santos, R., et al., Association of Postural Tachycardia Syndrome and Ehlers-Danlos Syndrome with Mast Cell Activation Disorders. Immunol Allergy Clin North Am, 2018. 38(3): p. 497-504.
- Seneviratne, S.L., A. Maitland, and L. Afrin, Mast cell disorders in Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet, 2017. 175(1): p. 226-236.
- Henderson, F.C., Sr., et al., Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet, 2017. 175(1): p. 195-211.
- Henderson, F.C., Sr., et al., Atlanto-axial rotary instability (Fielding type 1): characteristic clinical and radiological findings, and treatment outcomes following alignment, fusion, and stabilization. Neurosurg Rev, 2021. 44(3): p. 1553-1568.
- Henderson, F.C., Sr., et al., Craniocervical instability in patients with Ehlers-Danlos syndromes: outcomes analysis following occipito-cervical fusion. Neurosurg Rev, 2024. 47(1): p. 27.
- Gensemer, C., Daylor, V., Nix, J., Norris, R. A., & Patel, S. Co-occurrence of tethered cord syndrome and cervical spine instability in hypermobile Ehlers-Danlos syndrome. Frontiers in Neurology, 2o24, 15. doi:10.3389/fneur.2024.1441866
- Henderson, F.C., et al., Deformative stress associated with an abnormal clivo-axial angle: A finite element analysis. Surg Neurol Int, 2010. 1.
- Marathe, N., Lohkamp, L. N., & Fehlings, M. G., Spinal manifestations of Ehlers-Danlos syndrome: a scoping review. J Neurosurg Spine, 2022, 37(6), 783-793. doi:10.3171/2022.6.Spine211011
- Marianayagam, N.J., et al., Increase in clivo-axial angle is associated with clinical improvement in children undergoing occipitocervical fusion for complex Chiari malformation: patient series. J Neurosurg Case Lessons, 2021. 2(23): p. Case21433.
- Carley, M. E., & Schaffer, J., Urinary incontinence and pelvic organ prolapse in women with Marfan or Ehlers Danlos syndrome. American Journal of Obstetrics and Gynecology, 2000, 182(5), 1021-1023. doi:10.1067/mob.2000.105410
- Patel, M., & Khullar, V., Urogynaecology and Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet, 2021, 187(4), 579-585. doi:10.1002/ajmg.c.31959
- Boileau, A., Brierre, T., Castel-Lacanal, É., Soulié, M., & Gamé, X., Lower urinary tract involvement in Ehlers-Danlos and Joint Hypermobility syndromes: Review of the literature. Fr J Urol, 2024, 34(13), 102698. doi:10.1016/j.fjurol.2024.102698
- De Wandele, I., et al., Dysautonomia and its underlying mechanisms in the hypermobility type of Ehlers-Danlos syndrome. Semin Arthritis Rheum, 2014. 44(1): p. 93-100.
- Brock, I., W. Prendergast, and A. Maitland, Mast cell activation disease and immunoglobulin deficiency in patients with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorder. Am J Med Genet C Semin Med Genet, 2021. 187(4): p. 473-481.
{kind=link}